
Composite structures can improve the physical properties of hydrogels, making them suitable for structural and high-performance applications. Incorporating stiff reinforcement into hydrogels is difficult, because the swelling ratio of hydrogels change in response to their environment, causing stress at the interface between the gel and reinforcement. Here we introduce methods to over overcome this problem, including the preferential use of 1) deswelling hydrogels, 2) adhesive hydrogels, and 3) reinforcing phases that can adjust their size in response to the swelling ratio of the hydrogel matrix. These hydrogels exhibit strength and toughness that far surpasses traditional tough hydrogels.

1. Fabric Reinforced Hydrogel Composites
Reinforcing polymers with fibers has long been a common method to increase the strength and toughness of mechanically poor materials. Traditionally, a stiff carbon or glass fibers were impregnated with a hard, thermosetting resin to form hard and inflexible structural materials. These types of composites have found use in a wide range of applications, from building materials to airplane wings. Interestingly, many biomaterials also possess composite structures, consisting of stiff reinforcing biomaterials, such as collagen fibrils, within a soft extracellular matrix.[1] Compared to synthetic fiber-reinforced polymers, the ratio in stiffness between the reinforcing phase and matrix is much greater. To attempt to create synthetic biomaterials, efforts have been made to create fiber-reinforced soft composites, by using hydrogels as a matrix. However, this has proven difficult, as the volume of a hydrogel can change with the surrounding environment and swelling or deswelling causes stress at the interface with the reinforcing phase. Because of these problems, there has been little success in developing robust fiber reinforced hydrogel composites.
1-1. Synergistic Toughening in Fiber Reinforced Polyampholyte Hydrogels
To overcome the problems introduced by swelling matrices, we developed hydrogel composites by using polyampholyte hydrogels as the matrix hydrogel.[2][3]Polyampholyte hydrogels, a type of tough, viscoelastic hydrogel, do not swell when placed into pure water; in fact, in some cases they slightly deswell, decreasing in volume by about 20%. Furthermore, polyampholyte hydrogels possess self-adjustable adhesive properties: when in contact with charged surfaces (such as the hydroxyl groups present on the surface of glass fibers) the internal structure of the polyampholyte can rearrange to form strong electrostatic interactions with the surface.[4][5] These beneficial properties allow us to overcome the shortcomings of traditional hydrogels when fabricating fiber reinforced hydrogel composites.[6]

The fabric reinforced hydrogel composites fabricated from polyampholyte hydrogels exhibit a synergistic increase in toughness, more than an order of magnitude greater than either of the neat components. This means that the composites exhibit extreme tear-resistance: while traditional gels can be easily broken with one’s fingers, and unreinforced fibers fall apart easily, fiber reinforced polyampholyte hydrogels are so tough that they cannot be torn by hand. Tearing energy values above 100,000 J/m2 have been achieved.[7] Despite this high toughness, these materials are still flexible, and can be bent and molded into complex geometries. Polyampholyte hydrogels contain about 50% water by mass, making these composites similar in water content to load-bearing tissues in the human body. Due to these unique properties, fiber-reinforced polyampholyte hydrogels may find use in applications such as biological prosthetics.
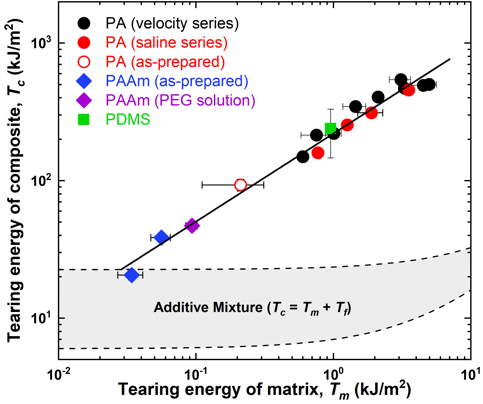
The mechanism for the synergistic increase in toughness is still being investigated. What is currently understood is that there is a direct correlation between toughness of the matrix and the resulting toughness of the composite. Because traditional hydrogels are brittle, when attempting to use them in composite structures, the resulting toughness cannot increase dramatically. We hypothesize that tough matrices allow for energy to be distributed to fibers far from the crack, which prevents the crack from growing. As the toughness of the matrix increases, we can create extremely tough composite structures. Despite having many similarities with biological soft tissues, by learning from these structures we have developed new synthetic materials with significant load-bearing capabilities.
Macroscale Double Network Composites
Double network hydrogels generally consist of two interpenetrating polymer networks: the first network is stiff and brittle, while the second network is soft and stretchable.[8][9] When combined, the resulting composite gel exhibits enhanced toughness, because the first network acts as a sacrificial scaffold, fracturing to dissipate energy and preventing crack growth. In traditional double network gels, both networks exist on the molecular scale. Here, our aim is to expand the length scale over which sacrificial bonds can be imagined. Demonstrating that sacrificial bonds and the “double network” concept works on length scales far beyond the molecular scale will open these materials for structural applications.
2-1. Low Melting Point Alloys as a Stiff and Tough, Healable Sacrificial Network
Due to the swelling mismatch between hydrogels that swell or deswell in the presence of water, and a rigid reinforcing frame that maintains a constant volume, creating hydrogel composites is difficult. Recently we developed a method to overcome this swelling mismatch problem, by incorporating low melting-point alloy (LMA) frames into bulk hydrogel structures.[10] LMA’s are metals that have melting points close to room temperature, and can easily be melted and reformed with a simple oven or hot water. A 3D printer was used to create specific designs that were then molded using silicone rubber. The melted LMA was injected into the silicone mold and cooled to room temperature to create specific designs. These LMA grids were then placed into a reaction vessel along with the hydrogel solution. After polymerization, a macroscale composite could be obtained. Depending on the type of hydrogel used, when placed into water the gel would either swell, taking up water from the environment, or deswell, releasing water to the environement. This process would cause the compsite structure to bend and buckle. At this point, the composite was placed in hot water, and the LMA melted, releasing the stress in the system. If the hydrogel swelled, additional LMA was injected to maintain the shape of the LMA grid. When the hydrogel deswelled, extra LMA was squeezed out of the hydrogel.

LMA composites made with a tough hydrogel matrix exhibits enhanced toughness compared to the neat hydrogel, due to interal fracture of the LMA structure. Importantly, this process could be cycled indefinitely. After fracture of the LMA grid, we placed the composite in hot water to melt the farctured grid, then cold water to reform it. These results demonstrate that not only is it possible to achieve the “double network effect” on the macroscale, we can achieve these properties in a way that is fully healable.Due to the incorporation of a metal frame, these composites have other unique physical properties. For example, they are electrically conductive. We used this property to perform electrolysis within the hydrogel. Also, temporarily heading and reshaping the structure allowed for shape-memory properties. Furthermore, the LMA could be melted and subsequentially removed to create hydrogel fluidic devices. Depending on the average size of the hydrogel network, these structures could be used for binary filtration of particles of varying sizes.
The use of LMA frames within hydrogel composites results in many unique mechanical and physical properties. Further demonstration of the macroscale double network effect is currently being persued.
References
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia, R. O. Ritchie,Nat. Mater. 2015, 14, 23.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima, J. P. Gong, Nat. Mater. 2013, 12, 932.
- A. Bin Ihsan, T. L. Sun, S. Kuroda, M. A. Haque, T. Kurokawa, T. Nakajima, J. P. Gong,J. Mater. Chem. B 2013, 1, 4555.
- C. K. Roy, H. L. Guo, T. L. Sun, A. Bin Ihsan, T. Kurokawa, M. Takahata, T. Nonoyama, T. Nakajima, J. P. Gong,Adv. Mater. 2015, 27, 7344.
- P. Rao, T. L. Sun, L. Chen, R. Takahashi, G. Shinohara, H. Guo, D. R. King, T. Kurokawa, J. P. Gong,Adv. Mater. 2018, 30, 1801884.
- D. R. King, T. L. Sun, Y. Huang, T. Kurokawa, T. Nonoyama, A. J. Crosby, J. P. Gong,Mater. Horizons 2015, 2, 584.
- Y. Huang, D. R. King, T. L. Sun, T. Nonoyama, T. Kurokawa, T. Nakajima, J. P. Gong,Adv. Funct. Mater. 2017, 27, 1605350.
- J. P. Gong, Y. Katsuyama, T. Kurokawa, Y. Osada,Adv. Mater. 2003, 15, 1155.
- J. P. Gong,Soft Matter 2010, 6, 2583.
- R. Takahashi, T. L. Sun, Y. Saruwatari, T. Kurokawa, D. R. King, J. P. Gong,Adv. Mater. 2018, 30, 1706885.